Quantum Mechanics - Module 3
Introduction
Many students are nervous of quantum mechanics, particularly the mathematics used, or think that it is irrelevant to chemistry and biology. In this module we hope you will discover that quantum mechanics is not much more difficult than other topics, that it does apply to the real world and, most importantly, that it is crucial to a fundamental understanding of physics, chemistry, and biology.
If the following picture is your mental image of an atom, with electrons looping around the nucleus, you are about 70 years out of date.
The modern world of quantum mechanics gives us the probability plots (shown below) for where an electron is most likely to be found in the various atomic orbitals of a hydrogen atom (the nucleus is at the center of each plot).
You have seen these plots for years already, but no one has probably ever explained why they should be this way electrons behave. It has just been something that you had to accept as Truth from the wise physicists. The aim of this unit is to reduce the element of faith in your understanding of atomic structure and increase the element of experimental understanding.
Understanding of some of the material in this Module can be assisted by taking advantage of a number of interactive Java applets available on the internet. These exercises are identified by the logo on the left, and will naturally require internet access. There will also be an opportunity at the Residential School to access this material through UNE’s network. Links to all exercises – and additional links – can be found at http://www.une.edu.au/chemistry/CHEM201_QM/CHEM_201.html.
Quantum chemistry applies quantum mechanics to problems in chemistry. The influence of quantum chemistry is felt in all branches of chemistry:
- Physical chemists use quantum mechanics to calculate (with the aid of statistical mechanics) thermodynamic properties of gases (e.g. entropy, heat capacity); to interpret molecular spectra, thereby allowing experimental determination of molecular properties (e.g. bond lengths and bond angles, dipole moments, barriers to internal rotation, energy differences between conformational isomers); to calculate molecular properties theoretically; to calculate properties of transition states in chemical reactions, thereby allowing estimation of rate constants; to understand intermolecular forces; and to deal with bonding in solids.
- Organic chemists use quantum mechanics to estimate the relative stabilities of molecules, to calculate properties of reaction intermediates, to investigate the mechanisms of chemical reactions, to predict aromaticity of compounds, and to analyze NMR spectra.
- Analytical chemists use spectroscopic methods extensively. The frequencies and intensities of lines in a spectrum can be properly understood and interpreted only through use of quantum mechanics.
- Inorganic chemists use ligand field theory, an approximate quantum mechanical method, to predict and explain the properties of transition-metal complex ions.
Although the large size of biologically important molecules make quantum-mechanical calculations on them extremely difficult, biological chemists are beginning to benefit from quantum mechanical studies of conformations of biological molecules, enzyme-substrate binding, and solvation of biological molecules.
This module will be divided into three topics: 3A – Pre-Quantum Mechanics, 3B – Introduction to Quantum Mechanics, and 3C – Model Systems and the Hydrogen Atom. These are derived from the chapter headings in the book ‘Physical Chemistry’ by David Ball, on which this module was originally based. The module will bring us to the gate of Quantum Chemistry, but will not truly take us through, because it is the quantum mechanics of things much more complicated than hydrogen atoms that chemists are mostly interested in. To a good approximation, however, the whole vast menagerie of molecules can be understood by putting together the basic concepts we will derive for the hydrogen atom.
Besides being useful, quantum mechanics can be an exhilarating mental exercise, as it is concerned with the behaviour of entities that are very very different from the macroscopic things we are used to dealing with. Along with these notes you will receive a copy of Chapter 5: The Naked Atom, from “The God Particle” (L. Lederman, Houghton Mifflin, Boston, 1992). On p. 143 Lederman writes:
“But be forewarned! The microworld is counterintuitive: point masses, point charges and point spins are experimentally consistent properties of particles in the atomic world, but they are not quantities we can see around us in the normal macroscopic world. If we are to survive together as friends through this chapter, we have to recognize hangups derived from our narrow experience as macro-creatures. So forget about normal; expect shock, disbelief. Niels Bohr, one of the founders, said that anyone who isn’t shocked by quantum theory doesn’t understand it. Richard Feynman asserted that no one understands quantum theory.”
Topic 3A
Pre-Quantum Mechanics
Towards the end of the nineteenth century many physicists felt that all the principles of physics had been discovered; little remained but to clear up a few minor problems and improve experimental methods to ‘investigate the next decimal place’. Those discoveries constitute what is now called classical physics. The early 20th century saw the introduction of the theories of relativity and quantum mechanics. Quantum mechanics was developed by several people over a period of several decades, and it is an extension of classical physics to subatomic, atomic and molecular sizes and distances. Relativity theory and quantum mechanics constitute what is now known as modern physics. Although relativity has had little impact in chemistry (why do you suppose this is the case?) quantum mechanics has played a very important role, so that an introductory course in quantum mechanics and its applications to chemistry - quantum chemistry- is an integral part of any chemistry course.
Laws of motion – classical mechanics
You might find it quite strange to see material on classical mechanics in a course on quantum theory, especially of quantum theory as relevant to chemistry. But it really is illuminating to revise and discuss several concepts in classical mechanics before embarking on a description of quantum mechanics.
Classical mechanics describes the motion of objects. To see how it does this, we will look at equations describing the constancy of energy and the response of particles to external forces.
Total energy
The total energy of a particle is the sum of its kinetic energy, K (the energy arising from its motion), and its potential energy, V (the energy arising from its position):
E = K + V
but recall that
K = ½ m v2 (m mass; v velocity) hence:
E = ½ m v2 + V
We can express this in terms of the linear momentum, p = mv:
p2/2m + V
Using the fact that v = dx/dt, we can rearrange this equation into a differential equation for x as a function of t:
E = ½m(dx/dt)2 + V
2(E - V) = m(dx/dt)2
Hence: dx/dt = [2(E - V] / m]2
This equation is important, because the solution for a given E yields x as a function of t. Substituting that value of x into the expression for V(x) yields the particle's velocity at that instant. Knowledge of x(t) and v(t) (or p(t)) enables us to describe the trajectory of the particle, and hence predict its location at any instant! This is the key to the success of classical mechanics.
Newton's second law – rectilinear motion
For motion in a straight line, this can be written in various forms:
F = ma = m dv/dt = m d2x/dt2 = dp/dt
(i.e. the rate of change of momentum equals the force acting on the particle). Hence if we know the force acting on the particle at all times and positions we can obtain its trajectory as above. As an example, consider a particle with initial momentum p(0)= 0 (i.e. at rest), subjected to a constant force F for time τ (Greek: “tau”), then allowed to move freely. Then we must have:
dp/dt = F(a constant) between t = 0 and t = τ
dp/dt = 0 > t > τ
The solution is p(t) = Ft, for 0 < t < τ
Since K = p2/ 2m, then the energy after the force ceases to act is E = F2 τ2/ 2m. Note that the particle is initially at rest, and F and τ can assume any value, which implies that (in classical mechanics) the energy of a particle undergoing rectilinear motion may be increased from 0 to any arbitrary value.
Rotational motion
In this case similar considerations apply, and rotational motion is a very important type of motion in chemistry (can you imagine why?). The concepts of momentum and velocity used above become angular momentum and angular velocity for rotational motion. The angular momentum, J, of a particle is J = Iω (compare this with p = mv) where I is the moment of inertia and ω (Greek: “omega”) the angular velocity. For a point particle of mass m moving in a circle of radius r, I = mr2.
To accelerate a rotation we need to apply a torque, T, or a twisting force, and Newton's equation becomes dJ / dt = T (compare this with dp / dt = F). As for rectilinear motion, application of a constant torque for time τ increases the rotational (kinetic) energy by E = T2 τ2/ 2 I. Again, the implication is that an appropriate torque and time interval can excite the rotation to an arbitrary energy. Classical rotational motion is reviewed in Section 18.7 of the textbook.
The harmonic oscillator
A third type of motion fundamental in chemistry is oscillatory, such as the vibration of atoms in a bond. A harmonic oscillator consists of a particle that experiences a restoring force proportional to its displacement from an origin:
F = -kx
where the negative sign indicates the force is opposite to the displacement, and k is called the force constant. Using Newton's second law we can write:
dp/dt = md2x/dt2 = F = -kx
This is a second-order differential equation, readily solved (check this by substituting the solution into the equation) to yield:
x = Asinωt
p = mdx/dt = mωt
ω = √k / m ⇒ mω2 = k
You should be able to see that the position of the particle varies harmonically (or sinusoidally) with a frequency ν = ω / 2π (ν is Greek “nu”), and is stationary
(dx/dt = 0 ⇒ p = 0 ⇒ ωt = nπ/2
when x has its maximum value of ±A, the amplitude of motion (i.e. at the extremes of its oscillation). The potential energy (yes, it must also possess potential energy, since its kinetic energy is clearly not constant - it is zero at x = ±A!) can be obtained from F = –dV/dx, hence V = ½kx2(if V = 0 at x = 0). Then the total energy can be written:
E = ½p2/ m + V
= ½p2/ m + ½kx2
= ½mω2A2cos2ωt + ½kA2sin2ωt
= ½kA2(cos2ωt + sin2ωt)
= ½kA2
From this we can see that the energy of an oscillating particle can also assume any arbitrary value. Note that the frequency of oscillation depends only on the structure of the oscillator (i.e. k and m) and not on its energy! Amplitude of oscillation determines energy, and it is independent of the frequency. The classical simple harmonic oscillator is reviewed in section 18.6 of the textbook.
The equipartition theorem
This useful theorem states that the average value of each quadratic term in the energy expression for a particle at temperature T is ½kT where k is the Boltzmann constant, k = 1.381 × 10-23 JK-1.
(The gas constant, R, is the product of k and NA, R = kNA).
It follows from the equipartition theorem that the average energy of a gas in a container (i.e. undergoing three-dimensional translational motion) is 3/2 kT (from E = (px2 + py2 + pz2)/ 2m ; the average energy of a collection of one-dimensional harmonic oscillators is kT (from E = ½p2/m + ½kx2), and the average energy of a collection of objects rotating about a single axis is ½kT (from ½Iω2 ).
Note that all of these expression for the total energy of a system are expressed in terms of momenta (for a three dimensional system, we have three separate momentum variables, px, py, and pz) and position (x,y,z). We will use the same variables when we move from classical to quantum mechanics.
The failures of classical physics
Read Engel and Reid
Chapter 12
Two important conclusions emerge from the classical descriptions of motion:
classical physics:
- predicts a precise trajectory, and
-
- allows all forms of motion to be excited to any energy simply by controlling the forces, torques etc. that may be applied.
It turns out that classical mechanics, which is excellent in describing everyday objects (ball bearings, planets, etc.) fails in describing very small systems and transfers of very small energies. It is only an approximation to the description of matter on the atomic scale. However, its failures (which were spectacular at the turn of the last century) provided the impetus for a completely novel approach to the description of matter, and we now examine some of these examples in turn.
Blackbody radiation
All bodies emit radiation when heated. As the temperature increases, the apparent colour of the body changes from red to white to blue. In terms of frequency, the emitted radiation goes from a lower frequency to a higher frequency as the temperature increases. Red is in a lower-frequency region of the spectrum (λ ≈ 700 nm; ν ≈ 4.3 × 1014 s–1) than is blue (λ ≈ 450 nm; ν ≈ 6.7 × 1014 s-1). The exact spectrum emitted depends upon the body itself, but an ideal body, which emits and absorbs at all frequencies, is called a blackbody, and its emitted radiation is called blackbody radiation. A good approximation to a black body is a cavity with a tiny hole (see Fig 12.1, p.277 E&R).
The amount of energy radiated by a black body as a function of its temperature is described by the Stefan-Boltzmann law:
M = σT4
where σ (Greek “sigma”) is the Stefan–Boltzmann constant = 5.67 × 10-8 J m-2 s-1 K-4 and T is absolute temperature.
As the temperature is raised, more light is emitted at shorter wavelengths (higher frequencies). Frequency (ν) and wavelength (λ, Greek “lambda”) are related to the speed of light (c) by:
λν = c
Classical physics could not explain the temperature dependence of blackbody radiation (Fig 12.2, p.277 E&R).
Many theoretical physicists attempted to derive this distribution function, but were unsuccessful. The best attempt, by Rayleigh and Jeans (equation 12.3, p.277), predicted that the emitted energy by the black body depended on 1 / λ4. This successfully predicted the fall-off in emitted radiation at longer wavelengths (low frequencies). However, it does not predict a maximum but instead predicts increasing emitted intensities at shorter and shorter wavelengths. This failure of classical physics was called the “ultraviolet catastrophe” because it predicted significant radiation in the short wavelength, UV portion of the electromagnetic spectrum. According to this theory objects should glow even at room temperature! Rayleigh and Jeans assumed that the energies of these "oscillators" could have any value at all (a sensible outcome of classical physics as we have seen above) and they used the expression for the average energy of a collection of one-dimensional harmonic oscillators we have given above, E = kT.
Max Planck (1900) was able to resolve the discrepancy between theory and experiment, but it required a rather revolutionary assumption. Like Rayleigh and Jeans, Planck assumed that the radiation emitted was due to the oscillation of electrons in the matter of the body. Planck, however, made the assumption that the energies of the oscillators had to be proportional to an integral multiple of the frequency,
E = nhλ
(12.4)
where h is a proportionality constant. Using statistical thermodynamic arguments, Planck derived equations similar to 12.5 and 12.7 and showed that excellent agreement with experiment could be obtained if h = 6.626 x 10–34 J s. This constant is of course now known as Planck's constant. For small frequencies it is straightforward to show that Planck's equation reduces to the Rayleigh-Jeans law, but the Planck distribution does not diverge at large ν.
An empirical (what does this word mean?) relationship known as Wien's law was known in the late nineteenth century:
λmaxT =2.90 × 10-5
Exercise 3.1
Attempt problem 12.1 on page 287 of E&R.
If you are keen, attempt problem 12.12.
From equation 12.7, setting dρ / dλ = 0, it can be shown that Planck's distribution predicts
λmaxT = hc/4.965k = 2.90 × m K
The theory of blackbody radiation is not abstract; it is used regularly in astronomy to estimate the surface temperature of stars. For example, from the electromagnetic spectrum of the sun measured in the earth's upper atmosphere λmax = 500nm, and from Wien's law we deduce that T = (2.90 × 10–3 m K)/(500 × 10–9 m) = 5800 K
The photoelectric effect
Examine the Planck distribution as a function of wavelength and frequency at a variety of temperatures:
http://www.oup.com/uk/orc/bin/0198792859/resources/livinggraphs/graphs/P711S01.html
For some time, Planck's derivation was regarded as a curiosity - it was felt that a suitable classical explanation, one that avoided Planck's crucial assumption, would eventually be found. However, in 1905 Einstein used precisely the same idea to explain the photoelectric effect.
The ejection of electrons from the surface of metals when exposed to ultraviolet radiation is known as the photoelectric effect. Its characteristics are:
- no electrons are ejected, regardless of light intensity, unless the frequency exceeds a threshold value characteristic of the metal;
-
- the kinetic energy of ejected electrons is linearly proportional to the frequency of the incident light, but independent of its intensity;
- even at low light intensities, electrons are ejected immediately if the frequency is above the threshold.
Exercise 3.2
Attempt problem 12.11 on page 287 of E&R.
If you are keen, attempt problem 12.20.
The kinetic energy of the ejected electrons can be measured by the potential needed to be applied to a negative electrode to just stop them - the stopping potential VS (½mev2 = -eVS). These observations are summarised in Figure 12.4, p.279 E&R.
These observations suggested that the photoelectric effect depends upon the ejected electron being involved in a collision with a particle-like projectile with sufficient kinetic energy to knock it out of the metal. If we suppose the particle is a photon of energy νh, the conservation of energy requires that
½mev2 = hν - Φ
where Φ is the work function of the metal - the energy required to remove an electron from the solid. If hν < Φ photoejection cannot occur. This simple relationship predicts that a plot of VS> against ν should be linear, with a slope of –h / e (see Fig. 12.4). Using the known value of e Einstein (in 1905) obtained a value of h in close agreement with Planck's value from blackbody radiation.
Wave-like behaviour of particles
Scientists have always had trouble describing the nature of light. In some experiments it shows definite wavelike character, but in others it behaves as a stream of particles (now called photons). In 1924 the French scientist Louis de Broglie reasoned that if light displays wave-particle duality, then matter, which is certainly particle-like, may also display wave-like character under certain conditions. Einstein had already shown from relativity theory that the momentum of a photon is given by p = h / λ. De Broglie argued that both light and matter obey the equation
λ = h / p
(12.11)
This equation predicts that a particle with mass m moving at velocity v will have associated with it a wavelength λ = h mv. For example, the de Broglie wavelength of a cricket ball (156 g) travelling at 151.2 kph (40 m s–1) is 1.06 × 10–34 m!! On the other hand, the de Broglie wavelength of an electron accelerated from rest through a potential of 1.00 kV is 3.88 × 10–11 m = 0.388 Å. The wavelength of such electrons corresponds to the wavelength of X-rays, and like X-rays such electrons are diffracted by crystalline material. This diffraction of electrons was first demonstrated by Davisson and Germer in 1925 and is now commonly used in electron microscopes.
The most dramatic experiment demonstrating the wave nature of electrons is the double-slit diffraction experiment: a beam of electrons fired through an arrangement of two parallel slits will give a diffraction pattern, as though the electrons going through the two different slits could interfere with each other just like classical light beams. This even happens if the electrons are fired through one at a time, suggesting that somehow, a single electron can go through both slits simultaneously! It is possible to carry out experiments to find out which of two slits an electron went through... but if this is done, the interference patterns disappear. Subsequent experiments have shown that other particles, such as protons, neutrons, and even helium nuclei also exhibit this bizarre behaviour.
The wave-particle duality is not obvious from our usual macroscopic observations. Most macroscopic particles have significant masses such that their de Broglie wavelength is infinitesimally small. Microscopic particles, such as photons and electrons are neither waves nor particles, but something else. They can appear to behave as waves or particles, depending on what sort of experiment we carry out. An accurate pictorial description of these particles’ behaviour is impossible using only the wave or only the particle concept of classical physics. The concepts of classical physics have been developed from experience in the macroscopic world and do not provide a proper description of the microscopic world.
Although both photons and electrons show an apparent duality, they are not the same kinds of entities. Photons always travel at speed c and have zero rest mass; electrons always have ν < c and a nonzero rest mass. Photons must always be treated relativistically, but electrons whose speed is not too high can be treated nonrelativistically.
Another important consequence of the wave nature of particles is what is called the uncertainty principle. The momentum of a particle is a function of its wavelength (12.11), but it is impossible to specify an exact wavelength for a localised wave: if a wave packet occupies only a particular bit of space, it must necessarily be composed of a sum of waves with a number of different wavelengths. Thus, if a particle actually is somewhere, rather than everywhere, there will be some uncertainty in its wavelength, Δλ. In the same way, unless it is described by the sum of an infinite number of waves, there will be some uncertainty in its position, Δx. It is possible to put numbers on this trade-off between momentum and position, which is done much later on in the textbook (pp.360-366) to give the expression
ΔpΔx ≥ h / 2
Exercise 3.3
The following summary by the famous physicist Richard P. Feynman (The Feynman Lectures in Physics, III-1.10) defines an important term which we will meet again. [An ‘event’ is, for instance, an electron arriving at a specific point on a detector. The ‘several alternative ways’ are, for instance, electrons passing through slit 1 or slit 2.]
- The probability of an event happening in an ideal experiment is given by the square of the absolute value of a complex number Φ which is called the probability amplitude:
P = probability, Φ = probability amplitude P = |Φ |2
- When an event can occur in several alternative ways, the probability amplitude for the event is the sum of the probability amplitudes for each way considered separately. There is interference:
Φ = Φ1 + Φ2
P = |Φ1 + Φ1 + Φ2|2
- If an experiment is performed which is capable of determining whether one or another alternative is actually taken, the probability of the event is the sum of the probabilities for each alternative. The interference is lost:
P = P1 + P2
P1 = |&Phi 1|2
P2 = |Φ 2|2
[Pick any function you like for Φ1 and Φ2 and create plots illustrating cases (2) and (3) for those functions.]
Atomic Spectra
A detailed analysis of the emission spectrum of hydrogen was a major step in the elucidation of the electronic structure of atoms. In 1885 a Swiss amateur scientist, Johann Balmer, showed that a plot of the frequencies of the lines in the visible/UV spectrum versus 1/n2 is linear. In particular Balmer showed that
ν = 8.2022 × 1014(1 - 4/n2) Hz (Hz = s-1)
where n = 3,4,5,... etc. It is customary now to write this in terms of 1 / λ instead of n. Reciprocal wavelength is denoted in the text by ṽ, measured in the standard unit cm–1 (these are non-SI units, and are called wavenumbers). We can then obtain the Balmer formula:
ṽ = 1/λ = R(1/22 - 1/n2), n = 3,4,5,...
There are series of lines just like the Balmer series in the UV and infrared regions of the spectrum. The Swiss spectroscopist Johannes Rydberg accounted for all the lines in the H atomic spectrum by generalising the Balmer formula:
ṽ = RH(1/n22 - 1/n21)
(12.13)
Exercise 3.4
Attempt problem 12.17 on page 288 of E&R.
This is the Rydberg formula and the constant occurring in it is now called the Rydberg constant, RH = 109,677.57 cm–1.
The fact that the formula describing the entire H atom spectrum is controlled by two integers is quite surprising. But integers play a special role in quantum theory, as we shall see.
Topic 3B – Introduction to Quantum Mechanics
In 1926 both Schrödinger and Heisenberg independently formulated a general quantum theory - a new kind of mechanics - capable of dealing with the wave-particle duality of matter. They used quite different mathematical formalisms but both represent different forms of what is now known as quantum mechanics. Schrödinger's mathematics (wave mechanics) involves differential equations and is more familiar to chemists; it is usual to use his equations as the basis of chemical applications of quantum mechanics - quantum chemistry.
When do we use Quantum Mechanics?
Read Engel and Reid
Chapter 13.1
This section attempts to give a more quantitative answer to the question ‘when do we use quantum mechanics?’ than, ‘when we’re talking about really small things’. Crucial to answering this question is the Boltzmann distribution, which you may possibly remember from first year. It is important that you learn this important classical expression for the partitioning of particles (or oscillators, or any collection of systems) between two energy states separated by an energy difference ΔE:
ni/nj = gi/gj e -[εi - &epsilonj]/kT
(13.2)
where ni is the numbers of atoms that have energy &epsiloni and nj is the number of atoms that have energy εj, gi and gj are the degeneracies of the energy levels- the number of possible ways of arranging the system to get to that energy. k is Boltzmann’s constant (8.314 JK–1 / Avogadro’s Number) and T is the temperature.
Basically, if εi - εj << kT, then quantum mechanical behaviour will be observed: otherwise, the system will display a more-or-less continuous distribution of energy levels and behave more-or-less classically.
Read Engel and Reid
Chapter 13.2 and 13.3
Classical Waves
Consider a uniform string stretched between two fixed points (e.g. a string on a violin). The displacement of the string from its equilibrium horizontal position can be described in terms of a wavefunction ψ(x,t): the amplitude of the displacement as a function of both position x and time t. Using classical mechanics it can be shown that ψ(x,t) satisfies the equation
d2ψ/dx2 = 1/v2 d2ψ/dt2(13.11)
where is the speed with which the disturbance moves along the string and the other quantities are partial derivatives. This is called the classical non-dispersive wave equation. Solutions to this equation can be written as trigonometric functions, or as complex functions- you might remember Euler’s equation, eia = cosa + isina (if only for the gee-whiz expression, eip = –1).
Exercise 3.5
It can readily be shown that a sine wave, travelling in the x direction with velocity v, wavelength λ, frequency ν and amplitude A, described mathematically by
ψ(x,t) = A sin2π/λ< (x - vt) = A sin 2π(x / λ - νt)
is a solution to the wave equation 13.11. Prove this for yourself by substitution and differentiation – but note the difference in this equation between velocity v (which appears in the x – vt term) and frequency ν (which appears in the x/λ – νt term).
Read Engel and Reid
Chapter 13.2 and 13.3
Many problems in physics and chemistry are independent of time (e.g. the electron distribution or the energy of a molecule), and we can actually obtain a wave equation that does not contain t as a variable. In mathematics this procedure is known as separation of variables, and it is performed by assuming the solution ψ(x,t) can be written as the product of two functions, one of which depends only on x, and the other only on t:
ψ(x,t) = ψ(x)φ(t)
Substituting this form of ψ (x,t) into the classical wave equation results in:
Left hand side: ( ∂2ψ/∂x2 )t = φ(t)( d2ψ(x)/dx2 )
Right hand side: 1/v2 ( ∂2ψ/∂t2 )x = 1/v2ψ(x) ( d2φ(t)/dt2 )
dividing both sides by ψ(x)φ(t) we obtain
1/ψ(x) d2ψ(x)/dx2 = 1/v2 1/&phi(t) d2&phi(t)/dt2
function of x only function of t only
Since x and t are independent variables, the only way a function of x and a function of t can be equal for all x and t is if they both equal a constant, say, -β2 (this curious choice will become clearer below!). Then equating each side of the equation to -β2 results in two equations, one in x and the other in t.
Our interest is in the x-dependent equation (i.e. the part that is independent of time). The equation in x is:
1/ψ(x) d2ψ(x)/dx2 = -β2
and hence we get
d2ψ(x)/dx2 + &beta2ψ(x) = 0
This is a time-independent classical wave equation. It is easy to solve, because all we have to do is take the β2sinψ(x) back over to the right hand side and then look for a function that, when we differentiate it twice, we get the same function again times -β2. One solution would be sinβx, and another (remember Euler again) would be eβi...
The solution ψ(x) = Asinβx is equivalent to the time-dependent solution in Exercise 3.5 if we set t = 0 (check this for yourself). Hence we can identify β with 2π/λ, and we can then rewrite the time-independent wave equation in the form:
d2ψ(x)/dx2 + 4π2/λ2 &psi(x) = 0(13.17)
This differential equation is in precisely the same form as the key equation for quantum mechanics, the Schrödinger wave equation, and in fact we will use it to (sort of kind of) derive the Schrödinger wave equation. The quantum treatment of any chemical problem begins by setting up appropriate boundary conditions for this wave equation. The solutions ψ(x) of the wave equation are known as wavefunctions: they contain all the information we can hope to learn about the problem. That information can be retrieved by performing certain mathematical operations on the wavefunction. These three steps:
- setting up the wave equation;
- solving it to obtain the wavefunction;
- using the wavefunction to obtain energies, momenta, etc...
are standard procedures for any quantum mechanical problem.
Read Engel and Reid
Section 13.4
The Schrödinger equation
This section of the text derives Schrödinger's equation by assuming that atomic and molecular particles obey the time-independent wave equation, and that the wavelength of the particles obeys the de Broglie equation, λ = h/p.
Recall that we can write the total energy, E, as a sum of kinetic and potential terms:
Using p2 = (mv)2 = 2mK = 2m(E - V), we obtain p = [2m(E - V)]½, so that using the de Broglie relation λ = hp yields
λ2 = h2/2m(E - V)
Substituting this into the time-independent wave equation (13.17) we get
d2ψ(x)/dx2 + 4π22m(E - V)/h2 ψ(x) = 0
which is readily rearranged to obtain the Schrödinger equation for a particle of mass m moving in one dimension with energy E:
-h2/8&pi2m d2ψ(x)/dx2 + Vψ(x) = Eψ(x)
The Schrödinger equation can be rewritten in a more compact form:
H2/2m d2ψ/dx2 + Vψ = Eψ(13.20)
where H = h/2π (pronounced “h-bar”) and V and ψ are both functions of x. The first term in this expression can be identified with the kinetic energy of the system, and it has a similar form for all systems. But the potential energy term depends greatly on the system; for a free particle V = 0, and for a harmonic oscillator V = ½kx2
An interesting insight into the relationship between the form of the wavefunction ψ and the kinetic energy K can be obtained from
λ = h/(2m(E - V))½
= h/(2mK)½
Hence the greater the kinetic energy K, the smaller the wavelength. But we can relate the second derivative d2ψ/dx2 to the curvature of the wavefunction (i.e. the rate of change of slope). When a wavefunction is sharply curved (has lots of squiggles) the kinetic energy is large. When ψ is not so sharply curved (long wavelength, few squiggles) K is small. The association of curvature with kinetic energy is often a valuable clue to the interpretation of wavefunctions.
Read Engel and Reid
Section 13.5
Operators and observables
The Schrödinger equation can be rewritten in the more succinct form
(Ĥψ = Eψ)
(13.29)
where Ĥ is now an operator, something that operates on the wavefunction ψ(x). In this case we have
Ĥ = - η2/2m d2/dx2 + V
or in words, stepwise, take the second derivative of ψ, multiply it by H2/2m, and add to it the product of ψ. The operator Ĥ is a special one in quantum chemistry and is called the Hamiltonian operator.
The Schrödinger equation Ĥψ = Eψ is what is known as an eigenvalue equation:
(operator) × (function) = (constant) × (function).
Symbolically, these special equations are of the form
Ôf = ωf
where Ô is the operator, ω is the eigenvalue of the operator Ô (e.g. E is the eigenvalue for Ĥ) and f is the eigenfunction corresponding to that eigenvalue. As another example, eax2 is an eigenfunction of d/dx, but eax2 is not an eigenfunction of d/dx.
Exercise 3.6
Attempt problem 13.10 on page 308 of E&R
If you are keen, attempt problem 13.16.
If you are very keen and have lots of time on your hands, attempt all problems 13.10-13.20.
Eigenvalue equations are important in quantum mechanics because they are repeated for many other properties apart from energy, properties that in quantum mechanics are called observables. In general we can write
(operator) × (wavefunction) = (observable) × (wavefunction).
Therefore if we know the wavefunction and operator we can predict the outcome of an observation of that property by picking out the factor ω (the eigenvalue) in the eigenvalue equation.
Table 14.1, p. 313 of E&R, gives a convenient summary of operators for various observables (such as position, momentum, energy), along with their classical counterparts.
Read Engel and Reid
Sections 13.6 and 13.7
Properties of the wavefunction
One of the basic assumptions in quantum mechanics is that the state of a system (e.g. an electron, proton, atom, molecule….) can be described by its wavefunction, in just the same way that the displacement of a macroscopic string can be described by the wavefunction ψ(x,t).. Moreover, the wavefunction contains all the information we need to know about the system! And just as in classical mechanics, not just any old mathematical function will do – it must satisfy certain constraints in order to be physically reasonable.
The mathematical constraints include:
- ψ(x) and dψ(x) / dx must be everywhere finite;
- ψ(x) and dψ(x) / dx must be single-valued everywhere;
- &psi(x) and dψ(x) / dx must be continuous everywhere.
Exercise 3.7
Attempt problem 13.26 on page 309 of E&R
If you are keen, attempt problem 13.28.
Some examples of acceptable and unacceptable wavefunctions are given in Fig. 14.2, p. 312 of E&R. It is always advisable to keep these requirements in mind when seeking solutions to the Schrödinger equation: they are the key to physically correct solutions.
Read Engel and Reid
Section 14.1
The Born interpretation of the wavefunction
Assume that we can solve for ψ for a particular problem. What does it mean? In what ways can we learn about the properties of the particular system, be it the H atom, water molecule or a complex organic dye?
The answer to these questions, suggested by Max Born in 1926, showed that a wavefunction gives a description of a particle in terms of probabilities. Specifically, for a one-dimensional system, the probability of finding the particle in the region of space from x to x + dx is given by ψ2(x)dx (often this is written allowing for the complex nature of ψ - for example ψ(x) = Ae2x - as a product of ψ and its complex conjugate ψ*).
Thus ψ2 is a probability density (density, since it must be multiplied by infinitesimal length dx to get a probability), and ψ itself is called a probability amplitude.
We can then write down the probability of finding a particle (described by the real wavefunction ψ(x)) in a particular region of space, say between x = a and x = b, a < b,
P(a ≤ x ≤ b) = ∫ab&psi2(x)dx
i.e. the sum over all infinitesimal probabilities P(x)dx over the region.
Born's interpretation imposes very important constraints on the nature of the wavefunction, and it should now be clear why wavefunctions must be bounded (i.e. not reach infinity), single-valued, and continuous. A very important condition on a physically meaningful wavefunction is that of normalisation. Clearly, if we extend the limits of integration to (in words, over all space), the integral must equal unity: the particle must exist somewhere!
∫-∞∞ψ2(x)dx = 1(14.2)
Wavefunctions obtained from the Schrödinger equation will always have a multiplicative constant, N, the normalisation constant.
Read Engel and Reid
Sections 14.2, 14.3, and 14.4
The expectation value
This section of the text introduces the operators that are the quantum mechanical analogue of a collection of properties of the system, or observables, that are familiar to us from classical mechanics. There are two things we might want to do with these operators: find the result of an individual measurement, and find the average value we would obtain, if we carried out a large number of measurements. More often, we want to find the average value, since the general eigenvalue equation Ôf = ωf will have an infinite number of solutions ω.
In quantum mechanics the average value of an observable A whose operator is  is called the expectation value and is written like so:
<A>
It is calculated from
<A> = ∫ ψ ψdτ/∫ ψ2dτ
(14.4)
where dτ indicates integration over the appropriate variable(s). If the wavefunction ψ is normalised, than the bottom term of the expression is clearly equal to 1. The meaning of the integrand is that ψ is operated on by  to give a new function, and then this function is multiplied by ψ. It follows from this expression that, in the special case of the wavefunction ψ being an eigenfunction of the observable A with eigenvalue a:
<A> = a.
Read Engel and Reid
Sections 15.1 and 15.2
Translational motion: the particle in a box
Unfortunately, there are only a few systems for which solutions of the Schrödinger equation are straightforward and analytic (i.e. have a convenient mathematical form giving an equation that can actually be solved). These systems include (in order of increasing complexity): the free particle, the particle in a box, the harmonic oscillator, the rigid rotor, and the hydrogen atom. We will investigate each of these in turn.
Exercise 3.7
Attempt problem 15.2 on page 333 of E&R
If you are keen, attempt question 15.1.
The Schrödinger equation for free translational motion in one dimension is
-H2/2m d2ψ/dx2 = Eψ(i.e. V = 0)
It is easily shown that general solutions to this equation are of the form
ψ = Acoskx + Bsinkx
All values of k are permitted, hence all energies are allowed.
We are now ready to consider the problem of a particle in a one-dimensional box: a particle of mass m confined between two walls at x = 0 and x = a (see Fig. 15.1 p. 321 E&R). The potential energy is zero inside the box but rises abruptly to infinity at the walls. The Schrödinger equation for the region between the walls (where V = 0) is just that for a free particle, so the general solutions are the same as for a free particle.
Because the particle is restricted to the region (0, a) the probability that the particle is found outside this region is precisely zero. Hence ψ(x) = 0 for x outside (0, a). Mathematically, this is how we restrict the particle to the confines of the box. But to be an acceptable solution ψ(x) must be continuous and therefore ψ(0) = ψ(a) = 0, since any other choice would yield a discontinuity at the walls of the box. These are two boundary conditions on the wavefunction, which have dramatic implications for the form of the solution.
Using ψ(0) = 0 we must conclude that A = 0, since sin0 = 0, cos0 = 1. Hence our most general wavefunction must be of the form:
ψ(x) = B sin kx
But the amplitude at the other wall must also be zero, hence
ψ(a) = B sin ka = 0
This can have two solutions: either B = 0 (implying ψ = 0 for all x, a true but useless solution- such solutions are called trivial) or k must be chosen such that sin ka = 0 latter condition is satisfied provided
ka = nπ,n = 1, 2, 3, ...
Also, since k and E are related, we obtain
E = k2H2/2m = (nπ/a)2H2/2m = n2h2/8ma2,n = 1,2,3,...(15.17)
You should recognise immediately that the energy of a particle in a box is quantised – it can only take on certain values - and that this quantisation arises directly from the boundary conditions that ψ must satisfy to make it an acceptable wavefunction. The integer n is known as a quantum number. We can impose normalisation on the solutions to solve for the amplitude, B:
1 = ∫-∞∞ &psi2dx
= B2∫0a sin2 kxdx
= B2½∫0a (1 - cos2kx) dx
= ½B2[x - 1/2k sin 2kx]0a
= ½B2[a - 1/2k sin2ka - 0 + 1/2ksin 2k0]
= ½B2a
∴ B = √2/a
From this we can see that the wavefunctions are of the form:
ψn(x) = √2/a sinnπx/a,n = 1,2,3,...(15.15)
where we have labeled the wavefunctions with the quantum number n.
The wavefunctions are the same as standing waves set up in a vibrating string; the energy increases with the number of nodes.
(The energy levels can also be derived independently by observing that the wavelength is directly related to the size of the box via a = nλ / 2 (i.e. an integral number of half wavelengths)) and using this information in the de Broglie relation).
Exercise 3.8
Attempt problem 15.5 on page 333 of E&R
Explore the particle in a 1-D box:
http://www.falstad.com/qm1d/
Several properties of the particle-on-a-box solutions yield considerable insight. For example the average momentum of the particle in a box is zero; actually measurement of the linear momentum would give the value k half of the time, and –k the other half of the time. In other words this is the quantum mechanical version of the classical picture where the particle in a box rattles around, travelling to the right half the time and to the left the rest of the time. Examine also example 15.4, pp.331-332 of E&R which demonstrates that the average value of the position operator (i.e. the most likely position of the particle in the box) is simply the middle of the box:
<x> = a/2
Exercise 3.9
Examine section 16.13 on page 339 of E&R. Then attempt problem 16.3 on p.352.
Another application of the particle in a box solution is to the colour of polymethine dyes, and this is the subject of one of the experiments you will perform during this course
It is worthwhile examining carefully the plots of ψ2 for the particle in a box, Fig. 15.3, p. 323 of E&R, and especially comparing them with plots of the wavefunction in Fig. 15.2. ψ2 is a probability density, and as such is positive everywhere – unlike the wavefunction. The behaviour of ψ2 for large values of quantum number n shown in Figure 15.4 (p.324), and the approach towards the classical solution, is also important to note.
Remember that n ≠ 0 (since this implies ψ(x) = 0 everywhere) and hence the lowest energy that the particle may possess is not zero (as it would be classically) but E1 = h2 / 8ma2. This lowest energy is called the zero-point energy; its existence is a quantum mechanical effect, detectable only for small masses. If you like, it is a consequence of the uncertainty principle (Chapter 17): since the particle's location is not completely indefinite then its momentum cannot be zero, hence it must possess a finite k (kinetic energy). All of this because the particle is confined!
The particle in a three-dimensional box: degeneracy
Read Engel and Reid
Section 15.3
We can easily extend the results for one-dimension to the case of a three-dimensional box of sides a, b and c. The potential V is zero everywhere in the box and the Schrödinger equation is
-H2/2m ∇2ψ = Eψ
where we have introduced the Laplacian operator ∇2 :
∇2 = ∂2∂x2 + ∂2/∂y2 + ∂2/∂z2
The text describes how separation of variables can again be used to obtain the 3-D wavefunction in terms of a product of three 1-D wavefunctions. The solution is:
ψnx, ny, nz (x,y,z) = √8/abc sin (nxπx/a) sin (nyπy/b) sin(nzπz/c)
(15.24)
which makes it clear that each state of the system now requires specification of the three quantum numbers, nx, ny and nz.
In the same way that the wavefunction is a product of 1-D wavefunctions, the allowed energy levels are just sums of 1-D energies:
Enxnynz = h2/8m (nx2/a2 + ny2/b2 + nz2/c2)
(15.25)
If the box is a cube with side a, the energy expression becomes:
Enxnynz = h2/8ma2 (nx2 + ny2 + nz2)
(10.25)
Explore the particle in a 2-D box (especially useful for understanding the nature of wavefunctions and degeneracy):
http://www.oup.com/uk/orc/bin/0199280959/01student/graphs/graphs/lg_09.16.html
This is a fascinating result, as it clearly shows the existence of a new feature in quantum systems- the degeneracy we foreshadowed in our introduction to the Boltzmann distribution: more than one wavefunction may correspond to the same energy. For example, the three eigenfunctions ψ112, ψ121 and ψ211 correspond to different spatial distributions but they all have the same energy, equal to 6h2/8ma2. This energy level is said to be threefold degenerate. Many of the energy levels for a particle in a cubic box are degenerate – degeneracy is extremely common in highly symmetric systems. It is a direct consequence of the symmetry of the system, and many examples (such as the H atom) will be encountered later.
Tunneling
Read Engel and Reid
Section 16.5
A particle in a box is the simplest quantum mechanical problem to solve, and has been discussed in detail. Generalisation to a box of finite, rather than infinite depth is fairly straightforward, and is done in Section 16.1 (pp.337-338). This makes relatively little difference to the energies calculated for relatively high energy barriers, which is why the simple particle-in-a-box model we have derived can be used to give reasonable answers to physical problems. Where walls that are infinite in height, the particle cannot escape: its wavefunction is always zero outside the box. In real systems, however, the heights of the barriers are always finite, and the wavefunction does not have to be zero at the boundaries. Classically, the particle can only escape if its energy is equal to or greater than the energy of the walls. However, quantum mechanics predicts that the particle can escape even if its energy is less than the height of the walls: the wavefunction has a nonzero value outside the walls. The particle is said to tunnel through the barrier (see Fig. 16.7, p. 342 of E&R).
Exercise 3.10
Only if you are keen to exercise your mathematical muscles: attempt problem 16.6 on p.353. This problem is the only part of the text that goes into a quantitative understanding of tunneling.
A quantitative understanding of tunnelling is not expected for this course. However, the probability of tunneling depends on the energy and mass of the particle and on the height and width of the barrier. The mass of the particle is important because the de Broglie wavelength of a particle is inversely proportional to its momentum, mv. For constant kinetic energy, the wavelength of a wavefunction increases as the mass of the particle decreases: the longer the incident wavelength the higher the probability of transmission through the barrier. The wavelength of a 20 kJ mol-1 electron is 27 Â; the wavelength of a proton with the same energy is only 0.63 Â, so you can see that tunneling becomes negligible for larger particles. Quantum mechanical tunneling can have a major effect on the rates of reactions involving protons, and an even larger effect on electron transfer. Some applications of quantum tunnelling that illustrate it is not just a ‘gee whiz’ quantum oddity are given in sections 16.6 and 16.7.
The Uncertainty Principle and Entanglement
Read Engel and Reid
Chapter 17
The uncertainty principle is not really that peculiar: as we have mentioned before, it is just a straightforward consequence of the wave nature of particles. What is really peculiar is how a system ‘decides’ whether it is going to exhibit wave or particle behaviour when we experiment on it. The general rule is that if particles are indistinguishable from one another, they will interact like waves (P = |φ1 + φ2|2): if they can be distinguished, even in principle- whether we do it or not- they will interact like particles (P = |φ1|2 + |φ2|2). Have a look at the supplemental sections 17.5 and 17.6. Remember that many great physicists, such as Albert Einstein and Richard Feynman, have rejected the Copenhagen interpretation (p.368). In saying that a wavefunction ‘collapses’ when it is observed, are we giving the wavefunction a physical reality greater than it really has (remember, wavefunctions have imaginary parts!)? Are we confusing two layers of probability when we say that because the outcome of an experiment on identically prepared quantum systems is inherently probabilistic, we can’t say that the particle really is in one or box or another until we open it? Is the extreme weirdness of quantum mechanics entirely because the universe is really weird, or do some of these weird things appear simply because we don’t understand what is going on yet?
Exercise 3.11
Attempt problem 17.6, p.373 E&R.
Countless pages have been written about the philosophical implications of quantum mechanics, but these are really of no concern to us. We want to study how the rules of quantum mechanics gives rise to experimentally observable phenomena at the macroscopic level- the observable properties of chemical substances- and we will not bother with why they work or what they really mean. Because nobody knows. Quantum mechanics fits experimental data and predicts the results of experiments that have not yet been done better than any other model for chemical systems. And that is what we will do with it!
“One singular deception of this sort ... is to mistake the sensation produced by our own unclearness of thought for a character of the object we are thinking. Instead of perceiving that the obscurity is purely subjective, we fancy that we contemplate a quality of the object which is essentially mysterious; and if our conception be afterward presented to us in a clear form we do not recognise it as the same, owing to the absence of the feeling of unintelligibility.”
- Charles Sanders Peirce, ‘How to Make Our Ideas Clear’
Topic 3C – Model Systems
Synopsis
We have already covered the particle in a box, which is a model for the movement of electrons in conjugated systems, and for the translational motion of gases, and now we move on to model systems for other chemical entities. This topic covers the three most important model systems usually considered in a course of this kind: the harmonic oscillator (a model for vibrational motion), the rigid rotor in three dimensions (a model for rotational motion of molecules) and the hydrogenic atom. Hydrogenic atoms (i.e., anything with one nucleus and one electron, so He+, Li2+, U91+, etc.) are the only ones for which an analytical solution of the Schrödinger equation is possible. The solutions obtained (atomic orbitals and energy levels) form the basis for many of our more advanced ideas of the electronic structure of many-electron atoms and molecules.
The harmonic oscillator
Read Engel and Reid
Section 18.1
We saw earlier that an entity will undergo harmonic motion if it experiences a restoring force proportional to its displacement:
F = -Kx
where k is the force constant. We can rewrite this equation in terms of potential energy V rather than force F, which gives
V = ½kx2
(18.1)
Thus the Schrödinger equation for the system is
-h2/2m d2ψ/dx2 + ½kx2ψ = Eψ
(18.2*)
This version of the expression is slightly different from the one in the text: it assumes that our oscillating mass is attached by a ‘spring’ to something that doesn’t move at all, and hence doesn’t contribute to the equation of motion. There are similarities between the harmonic oscillator and the particle in a box. The particle undergoing harmonic motion is trapped in a symmetrical well where the potential rises to large values for large displacements. Again, boundary conditions must be satisfied, so we would expect the energy of the particle to be quantised. The solution of the Schrödinger equation is straightforward, but not simple. Rather than go through the derivation in detail, we will concentrate on a discussion of the solutions.
The most important feature of the result is that the permitted energy levels of a simple harmonic oscillator depend on a quantum number, n:
En = (n + ½) hvn = 0, 1, 2...
(18.6)
where the classical frequency of the oscillator is given (as earlier) by ν = (2π)-1√k/m.
The separation between energy levels is
ΔE = En+1 - En = hv
the same for all integers n, and hence the energy levels of the quantum mechanical harmonic oscillator are equally spaced (see Fig. 18.2, p. 381). This energy separation is negligibly small for macroscopic objects with large mass, but of great importance for objects of atomic masses. For instance, the force constant of a typical chemical bond is ~ 500 N m-1 and the mass of a proton is ~ 1.7 × 10-27 kg, hence ν ≈ 9 × 1013 s-1. The separation between adjacent energy levels is hν ≈ 6 × 10-20 J, which corresponds to 34 kJ mol–1, a chemically significant quantity. The radiation that is required to excite this transition must have &lambda = c / ν = hc / ΔE ≈ 3000 nm, in the infrared region.
The smallest value of n, the vibrational quantum number, is zero, hence a harmonic oscillator has a zero-point energy,
E0 = ½hv
As for the particle in a box, physically this must be the case because the particle is confined to a potential well, and therefore its kinetic energy cannot be zero.
The wavefunctions for the harmonic oscillator are interesting since they must resemble those for a particle in a box, but with very important differences. First, their amplitude falls to zero more slowly at large displacements since the potential well rises as x2, not infinitely steep. Second, since the kinetic energy K depends on the displacement in a complex way (i.e. it is not constant) the curvature of the wavefunction must vary in a more complicated manner. The wavefunction for the harmonic oscillator in a state with quantum number n is most generally expressed in the form
&psin = AnHn(√a x) e-αx2/2
where α is a constant (α is defined at the top of page 379 of Engel and Reid) An is a normalisation constant (also defined for all n at the top of page 379 of Engel and Reid), and Hn is known as a Hermite polynomial. The lowest-order Hermite polynomial is unity, hence the wavefunction for the ground state (lowest energy state) of the harmonic oscillator is
ψ0 = N0e-αx2/2..
and the probability density is just the bell-shaped Gaussian function,
ψ02 = N02e-αx2
Explore the nature of the wavefunctions as a function of quantum number:
Wavefunctions as a function of quantum number
which has its maximum at x = 0 (see Fig. 18.3, p. 381 of E&R). From Figure 18.4 it can be seen that at high quantum numbers harmonic oscillator wavefunctions have their largest amplitudes near the edges of the well - near the turning points of classical motion (where V = E, and K = 0!). Again, we see that classical behaviour emerges in the limit of large quantum numbers.
Example problem 18.2 (pp.379-380) gives far more mathematical detail than is necessary at 2nd year level, and certainly far more than you will be required to fully appreciate for this course. You will be expected to have a thorough understanding of the shape of the wavefunctions, the changes with quantum number, and the energy levels. We can now apply these solutions to the vibrational motion of diatomic molecules, and to the spectra we expect to observe: finally, you will see some of the chemical applications of quantum mechanics!
The simple harmonic oscillator is in fact a good (starting) model for a vibrating diatomic molecule. But how can this be? A diatomic molecule may be pictured as two particles (point masses) connected by a spring, but it does not look anything like a single mass experiencing a restoring force proportional to its displacement. It turns out that if we introduce centre-of-mass coordinates the two-body system can be reduced to a one-body system. This is a general result and not an approximation: If the potential energy depends only on the relative distance between two bodies, we can introduce relative coordinates and reduce the two-body problem to a one-body problem.
For a diatomic molecule we can define all motion relative to the centre-of-mass, and in this way obtain a quantity known as the reduced mass:
μ = m1m2/m1 + m2
The Schrödinger equation now becomes
(-h2/2μ) ( d2ψ/dx2) + ½kx2ψ = Eψ(ie. we use μ instead of m)
The resulting energy levels are still given by
En = (n + ½)hνn = 0, 1, 2...
where the vibrational frequency is now given by ν = (2π)-1 √k / μ . It can be shown (but not in this course!) that the harmonic oscillator model allows transitions between adjacent energy states only, so we have a selection rule
Δn = ±1 andΔE = En+1 - En = hν
so the observed frequency of the radiation absorbed (or emitted) is
νobs = 1/2π √k/μor
νobs = 1/2πc √k/μ, in terms of wavenumbers (in units of cm–1).
Exercise 3.12
Attempt problem 18.1 on p.401 of E&R.
If you are keen, attempt problem 18.15 as well.
The harmonic oscillator model therefore predicts that the spectrum of a diatomic molecule will be just one line, with frequency νobs, the fundamental vibrational frequency, and as we saw above, these transitions occur in the infrared region.
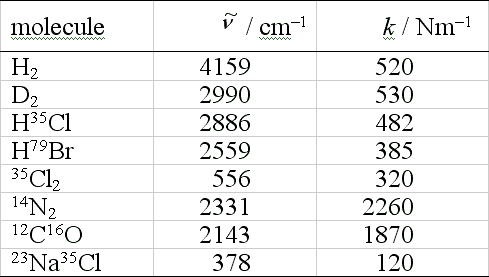
Fundamental vibrational frequencies and force constants for some diatomics are given in the table:
Rotational motion: the rigid rotor
Read Engel and Reid
Sections 18.2, 18.3 and 18.4
Another mode of molecular motion for which quantum mechanics provides important insights is rotation. For gas-phase molecules at low pressure, rotation is relatively free, at least during the time between collisions with other molecules or with the walls. For liquids and solids the model of a freely rotating molecule is not appropriate because, as soon as an individual molecule in a condensed phase begins to rotate, it bumps into its neighbors or experiences a restoring force that changes its direction. Such interrupted or irregular motion does not produce the sharply defined quantised energy levels characteristic of gas-phase molecules. In liquids and to some extent in solids, the rocking motions that do occur result in a broadening out or smearing of the sharp electronic and vibrational energies. As a consequence, the spectra of molecules in the condensed phase are usually much broader and lack fine resolution compared with gas-phase spectra. There are some exceptions in highly ordered crystals where molecules in a uniform environment may exhibit quite sharp spectra.
The treatment of rotational motion in Engel and Reid is rather more detailed than is necessary for this course. In particular, the detailed solution of the Schrödinger equation for two-dimensional rotational motion (Section 18.2) is not necessary for the course. Instead, we will focus on the broad details and, as before, on the nature of the solutions – the wavefunctions and energy levels. The wavefunctions are related to those for the hydrogen atom, and the energy levels tell us a great deal about rotational motion of diatomic molecules, and hence microwave spectroscopy, as you will see later.
The rigid rotator (or rigid rotor) is a simple model for a rotating diatomic molecule. The model consists of two point masses, m1 and m2 at fixed distances r1 and r2 from their centre of mass. Mathematically, this is the same as a single mass equal to the reduced mass μ, rotating about a point r = r1 + r2 from the mass. Note that although the diatomic molecule clearly vibrates as it rotates, it is a good first approximation to consider the internuclear distance as fixed.
We saw in beginning of this module that the kinetic energy of such a system is given classically by
K = ½ω2
where ω is the angular velocity and I is the moment of inertia, defined now by
I = μr2
The Hamiltonian operator for a rigid rotator contains just the kinetic energy term (since we can treat V as a constant, and hence for convenience set it to zero):
Ĥ = -ħ2/2μ∇2
Cartesian coordinates are no longer sensible (or simple to use) for a description of rotational motion in two or three dimensions. Instead, we use spherical polar coordinates, and it is important that you understand (not memorise!) how the Cartesian coordinates (x, y, x) are related to the spherical polar coordinates (r, θ, φ) (the angular variables are the Greek letters “theta” and “phi”).
The Laplacian in spherical polar coordinates can be written as
∇2 = ∂2/∂r2 + 2/r ∂/∂r + 1/r2 Λ2
where the angular operator is
Λ2 = 1/sin2 θ ∂2/∂φ2 + 1/sin θ ∂/∂θ (sin θ ∂/∂θ)
But because the particle is fixed to the surface of a sphere of radius r, there can be no term in Ĥ involving the partial derivative with respect to r. Therefore, the only term in ∇2 that remains is the last one, and
Ĥ = - ħ2/2μr2 Λ2
Using I = μr2, the Schrödinger equation simplifies:
- ħ2/2I Λ2ψ = Eψ
Notice that the moment of inertia, I, appears quite naturally in the denominator replacing m or m. This equation rearranges to give:
Λ2ψ = -2IE/ħ2 ψ
a partial differential equation in the two angular variables θ and φ. The solution is not particularly difficult, but you do not have to know its details for this unit. Briefly, the solution can be written as the product of two functions by separation of variables,
ψ(θ, φ) = Θ (θ) Φ (φ)
It can be shown that the acceptable wavefunctions are specified by two quantum numbers, l and ml, since there are two cyclic boundary conditions to satisfy, one arising from the angle φ and the other from θ. These quantum numbers are restricted to
l = 0, 1, 2, ...
ml = 0, ±1, ±2, ..., ±l(or ml = -l, -l + 1, ..., -1, 0, 1, ..., l - 1, l)
We see that l > 0, and ml depends on l: for a given value of l there are 2l + 1 permitted values of ml. The normalised wavefunctions are usually denoted Yl, ml (θ, φ) and are called the spherical harmonics.
The energy of the particle is restricted to the values
El = l(l + 1) ħ2/2I
(11.51)
from which we can see that the energy is quantised and independent of ml. Because of this, each level with quantum number l is (2l + 1)-fold degenerate.
As was the case for vibrational transitions, it can be shown that electromagnetic radiation can excite a rigid rotator (e.g. a rotating diatomic molecule) from one state to another. In particular, only transitions between adjacent states are allowed; the selection rule is
Δl = ±1
and in addition the molecule must possess a permanent dipole moment.
The energy difference between states is given by
ΔE = El+1 - El
= ħ2/2I [(l + 2)(l + 1) - l(l + 1)]
= ħ/2I 2(l + 1)
= ħ2 (l + 1)/I
= h2/4π2I (l + 1)
Using ΔE = hv, the frequencies at which absorption (or emission) may occur are given by
v = h/4π2 (l + 1)l = 0, 1, 2, ...
The reduced mass of a diatomic molecule is typically 10–25 to 10–26 kg, and internuclear separations are typically r ~ 10–10 m (1 Å), so I ~ 10–45 to 10–46 kg m2. Using I ~ 5 × 10–46 kg m2 gives absorption frequencies ν ~ 4 × 1010 Hz. These frequencies lie in the microwave region, hence rotational transitions of diatomic molecules lie in the microwave region, and comprise part of what is known as microwave spectroscopy.
It is common practice to write
ν = 2B(l + 1)l = 0, 1, 2, ...
where
B = h/8π2I(in Hz)
is the rotational constant of the molecule. It is also common to express the transition frequency in wavenumbers (cm–1), to obtain
ν = 2B(l + 1)andB = h/8π2cI(in cm-1)
It is clear from this analysis that the rigid rotator model predicts that the microwave spectrum of a diatomic molecule consists of a series of equally spaced lines with a separation 2B Hz or 2B cm–1. Hence, from the separation between absorption frequencies in the microwave region we can obtain B or 2B, hence I, and knowing the masses deduce r, the internuclear separation.
Exercise 3.13
Attempt problem 18.17 on p.401 of E&R.
If you are keen, attempt problem 18.24 as well.
For all but the lightest molecules, rotational-energy-level spacings are much smaller than those for typical vibrations. The rotational spacings are small compared with thermal energy kT at room temperature, which means that many rotational energy levels are well populated in gases under these conditions. This means that gas-phase molecules will be distributed over many rotational energy levels at room temperature. Because of the wide spacing of vibrational levels, most molecules will be in one, or only a few, vibrational levels at room temperature. The spacing of energy levels has important effects on the statistical interpretation of energy and entropy.
Chapter 19 of Engel and Reid considers the vibrational and rotational motion of simple molecules in more detail, and demonstrates the experimental utility of these quantum mechanical models. You do not need to know the material in Chapter 19 for this unit: it will be covered in the Symmetry and Spectroscopy component of the unit PHYS 301.
The hydrogen atom – solution of the Schrödinger equation
Read Engel and Reid
Sections 20.1, 20.2 and 20.3
A hydrogenic atom is not all that different from a rigid rotor in three dimensions. To model a hydrogen atom quantum mechanically, we are going to take our rigid rotor (where V = 0), make r a variable rather than a constant, and introduce a V that is dependent on r by the simple Coulomb relation that applies to macroscopic charged systems. Our solutions should therefore share some similarities with the solutions for the three-dimensional rotor. This simple model for hydrogen serves as a prototype for the description of more complex atoms, and therefore of molecules, because no analytical solution of Schrödinger’s equation is possible for any atom or molecule consisting of more than two fundamental particles.
If you think carefully you should remember having seen the results of a quantum mechanical treatment of hydrogen in 1st year chemistry. Here we will see the familiar orbitals and their properties emerge from the Schrödinger equation.
Picture the H atom as a proton fixed at the origin, and an electron of reduced mass μ (μ = memp/(me + mp)) interacting with the proton through a Coulombic potential,
V(r) = -e2/4πε0r(ε0 = 8.854 X 10-12 J-1C2m-1, the permittivity of free space)
(20.1)
The factor -4πε0 arises because we are using SI units; the charge on the proton is +e, and on the electron it is –e, and r is the separation between proton and electron. The system clearly has spherical symmetry, suggesting we use spherical polar coordinates. The Schrödinger equation for the H atom in these coordinates is:
-ħ2/2μ ∇2 ψ(r, θ, φ) + V(r)ψ(r, θ, φ) = Eψ(r, θ, &phi), and hence
ħ2/2μ [∂2/∂r2 + 2/r ∂/∂r + 1/r2 Λ2]ψ + V(r)ψ = Eψ
where Λ2 = 1/sin2 θ ∂2/∂φ2 + 1/sin θ ∂/∂θ (sin θ ∂/∂θ) as for the rigid rotor.
Again, we can solve this by separating variables, in this case separating the angular variables from the radial dependence:
ψ(r, θ, φ) = R(r)Y(θ, φ)
The two separated equations that result are:
1. Λ2 (θ, φ) = -l(-l + 1) Y(θ, φ)and
2. d2/dr2 [rR(r)] - 2μ/ħ2 V'(r)rR(r) = -2μ/ħ2 ErR(r)
(20.5)
V?(r), the effective potential, is given by V'(r) = -e2/4πε0r + l(l + 1)ħ2/2μr2.
(20.6)
The first of these is the angular wave equation (1.) and it is the same as the one solved earlier for the rotation of a particle in three dimensions. The cyclic boundary conditions are the same, hence the angular wavefunction is a spherical harmonic, Yl,ml(θ, φ), specified by the quantum numbers l and ml
ml = 0, ±1, ±2, ..., ±lwith l > 0.
But something new occurs here: l is also involved in the radial wave equation(2.), which will place a new constraint upon the permissible values of l, and hence ml. We won't go through the tedious steps of solving the radial equation; it will suffice to look at the solutions, which can be found for energies
En = - Z2e4μ/8ε02 h2 n2n = 1, 2, 3, ...
(20.7)
In this equation Z is the charge on the nucleus, which is 1 for the hydrogen atom, 2 for the He+ ion, etc. The corresponding radial wavefunctions depend on both n and l, and are of the form
Rn, l(r) = ρlLn, l(ρ)e-ρ / 2n = 1, 2, 3, ...,l = 0, 1, 2, ..., n-1
where ρ is proportional to r
ρ = 2μ/nmea0r
and the constant a0 is known as the Bohr radius:
a0 = 4πε0ħ2/mee2 = ε0h2/πmee2 = 0.529177 Å
The factor Ln, l (ρ) is simply a polynomial in ρ (and hence in r) known as an associated Laguerre polynomial (e.g. L3, 0 = 6 - 6ρ + ρ2).
Exercise 3.14
Attempt problem 20.2 on p.451 of E&R.
If you are keen, attempt problem 20.7
The full wavefunction is of course ψ(r, θ, φ) = Rn, l (r) Yl, ml(θ, φ). We see that the wavefunctions of the H atom depend on three quantum numbers: n, l, and ml; the lowest energy wavefunctions are given explicitly on pp.438-439 of Engel and Reid. (These expressions are valid only for the hydrogen atom, but can be made accurate for any hydrogenic atom such as He+, C11+, by replacing a0 with a0/Z, where Z is the charge on the nucleus, wherever it occurs).
Before discussing the wavefunctions (= atomic orbitals!) in some detail, it is worth emphasising that this quantum theory of the hydrogen atom predicts the experimentally determined emission spectrum, both its dependence on integers, and also the precise proportionality factor – the Rydberg constant.
The hydrogen atom wavefunctions
Read Engel and Reid
Sections 20.4, 20.5 and 20.6
In your studies in 1st year chemistry or physics you will have already covered some of the hydrogen atom solutions, and you should readily identify the wavefunctions with atomic orbitals, and the quantum number l with the orbital labels s, p, d etc. (see p. 439 of the text). Here we will revise some of these general details, as well as clarify some of the ideas behind the more common graphical representations of atomic orbitals.
The radial functions display several interesting characteristics, which can be deduced from the r-dependence of the wavefunctions (see Table on pp.438-439):
- s-orbitals (i.e. l = 0) have a finite, non-zero, value at the nucleus; all other orbitals have a node there;
- all atomic orbitals can be characterised by their nodal character (the number of times they cross the axis and change sign); e.g. the 2s orbital has one radial node, 2p orbitals have none, 3s has two nodes, 3p orbitals have one, etc.
The term orbital is used here in place of the classical mechanical term orbit - it is something that is probabilistic, rather than deterministic. In an orbit, the electron always occupies a position on a curve in space, and its position can in principle be calculated. In an orbital, we can only talk in terms of the electron density- which we can think of as the probability of finding an electron somewhere- which we can obtain from the square of the wavefunction and which is non-zero everywhere in the universe. When an electron is described by one of the complete wavefunctions, we say that it occupies that orbital.
All the orbitals of a given value of n are said to form a single shell of the atom. It is common to refer to successive shells by the letters:
Thus all orbitals with n = 2 form the L shell. Orbitals with the same n but different l values form the subshells of a given shell. These subshells are generally referred to by the letters:
Hence we can get a 3p subshell, or a 4d subshell, etc. Since the energy levels depend only on n, all except s-orbitals are degenerate, and in general the degeneracy of each shell is n2.
s orbitals
The lowest energy state of the H atom is the 1s orbital, for which
ψ1, 0, 0 = (1/πa03)½ e-r/a0
Clearly the 1s orbital is spherically symmetric, but ψ depends on distance from the nucleus, decaying exponentially from its value at r = 0 (what is that value – you can calculate it!); the most probable point at which the electron will be found is at the nucleus. All s orbitals are spherically symmetric, differing in the number of radial nodes. For instance the 2s orbital has a single node where
2 - r/a0 = 0i.e. r = 2a0
We see that as n increases, the average distance of the electron from the nucleus increases. This average value is the expectation value
<r> = ∫rψ2dτ
where dτ = r2dr sin θdθdφ. But the angular integration yields 1 because the spherical harmonics are normalised. Thus
<r>1s = &int0∞ r(R1, 0)2r2dr
and you should be able to show that this equals 3a0/2.
Similarly it can be shown that <r>2s = 6a0.
Many textbook plots of orbitals and wavefunctions can be confusing, and it is instructive to compare the probability density function, ψ2, with the radial distribution function, P = 4πr2ψ2. ψ2dτ gives the probability of finding the electron in a small volume element, dτ, whereas Pdr is the probability of finding the electron anywhere within a spherical shell of thickness dr at distance r from the nucleus (the volume of the shell is 4πr2 dr).
Exercise 3.15
Attempt problem 20.20 on p.452 of E&R.
Fig. 20.9 and 20.10 of the text illustrate the difference between ψ2 and P = 4πr2ψ2 for the ground state wavefunction of the H atom. The maximum in P gives the most probable radius at which the electron will be found; for a 1s orbital in hydrogen, 0, precisely the Bohr radius! For a 2s orbital, r = 5.2a0 (check that this is the case on the appropriate plot in Fig. 20.10, p.449 of E&R).
p orbitals
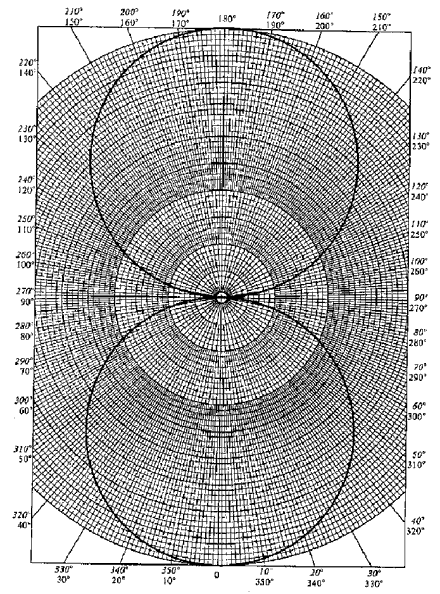
When l ≠ 0 the wavefunctions are not spherically symmetric; they depend upon both angular coordinates q and f, and it is therefore much more difficult to convey size and shape on a printed page. For l = 1 (p orbitals) ml = 0, ±1, hence there are three p-orbitals for each value of n. The angular part of the p-orbitals is given by the three spherical harmonic Y1, 0, Y1, +1 and Y1, -1. The simplest of these is
Y1, 0 (θ, φ) = √3/4π cos θ
Angular functions such as this can be expressed in the form of a polar plot on polar graph paper, where the value of cos θ, for example, is plotted along the radial line labelled by angle θ, as in the figure on the right.
A polar plot does not give the shape of the orbital (this must include Rn, l (r)), but it shows the direction or orientation of the orbital. In particular we can see that Y1, 0(θ, φ) is directed along the z axis, hence the orbital with angular dependence Y1, 0 is called a pz orbital.
Another common representation of orbitals is as 3-D figures. Since Y1, 0 is independent of θ, a 3-D representation can be <p>obtained from the polar plot by rotating it about the vertical axis in the figure.
The angular functions with ml ≠ 0 are more difficult to represent pictorially, as they not only depend upon q and f, but have imaginary parts as well:
Y1, +1 (θ, φ) = √3/8π sin θe+i, φand
Y1, -1 (θ, φ) = √3/8π sin θe-1, φ
Because Y1, +1 and Y1, -1 are degenerate, any linear combination of the two must also be a valid wavefunction with the same energy. It is customary to use the combinations given on p. 440 of the text which yield real wavefunctions.
3-D figures of all three real 2p orbitals are no doubt familiar to you- they feature prominently in the first chapters of most organic chemistry textbooks, for instance. Such plots show the surfaces of the orbitals within which the probability that the electron will be located is 90%, or some other high value. They are actually approximations to these 90% probability surfaces, and you will find quite different plots in various texts which claim to be representations of the same surface! Note that Engel and Reid do not actually give any 3-D figures: instead, in Figure 20.7, they show a two-dimensional contour plot of the 2py and some other orbitals. This is a good reminder that orbitals really don’t have ‘surfaces’- any atomic orbital you care to name is non-zero everywhere in the universe.
d orbitals

For the l = 2 case, ml =0, ±1, ±2 and so there are five d-orbitals. The simplest is the 3dz2 wavefunction, whose angular dependence is given by
Y2, 0(θ φ) = √5/16π (3 cos2θ - 1)
This angular part is presented as a polar plot in the figure on the right.
For other values of ml we can take linear combinations like we did for the 2p functions. The customary linear combinations are:
3dxz = 1/√2 (Y2, +1 + Y2, -1) = √15/4π sinθ cosθ cos φ
3dyz = 1/√2i (Y2, +1 + Y2, -1) = √15/4π sinθ cosθ sinφ
3dx2 - y2 = 1/√2 (Y2, +2 + Y2, -2) = √15/16π sin2θ cos 2φ
3dxy2 - y2 = 1/√2i (Y2, +2 + Y2, -2) = √15/16π sin2θ sin 2φ
The angular parts of all five real d orbitals are also given on p.441 of Engel and Reid. Note that the last four orbitals differ only in their orientations, although they look quite distinct mathematically.
Other representations of the various real s, p, and d orbitals include probability densities proportional to the density of dots (as in the Introduction to this Module):

The end and the beginning
The hydrogenic atom really is about the most complicated system for which an analytical solution is possible. For anything more complicated, we can only use numerical methods to get approximate values for the energies of the system. In the application of Quantum Mechanics to chemistry, we build up our approximate wavefunctions for molecules from combinations of these hydrogenic wavefunctions. So, in this module we have brought you only as far as the threshold of chemistry: we have stopped just after introducing the orbital shapes that are all familiar to you from the first chapters of your general chemistry textbooks! But now you should have a better idea of where they come from. If you want to pursue Quantum Chemistry further, you should enrol in PHYS 301 for third year.
Summary
This module provides an introduction to the formal theory of quantum mechanics and illustrates its use with applications to some simple systems. The following lists the new concepts that have been introduced. Ensure that you are familiar with all the points listed and revise appropriate sections where necessary.
- The origins of quantum mechanics; failures of classical physics, wave-particle duality.
- The dynamics of microscopic systems; Schrödinger equation, Born interpretation of the wavefunction.
- Quantum mechanical principles; information in a wavefunction, the uncertainty principle.
- Translational motion; particle in a box, motion in two or more dimensions, tunneling.
- Vibrational motion; energy levels and wavefunctions.
- Rotational motion; rotation in three dimensions; energy levels and wavefunctions.
- The structure and spectra of hydrogenic atoms; atomic orbitals and their energies; visualisation of atomic orbitals.